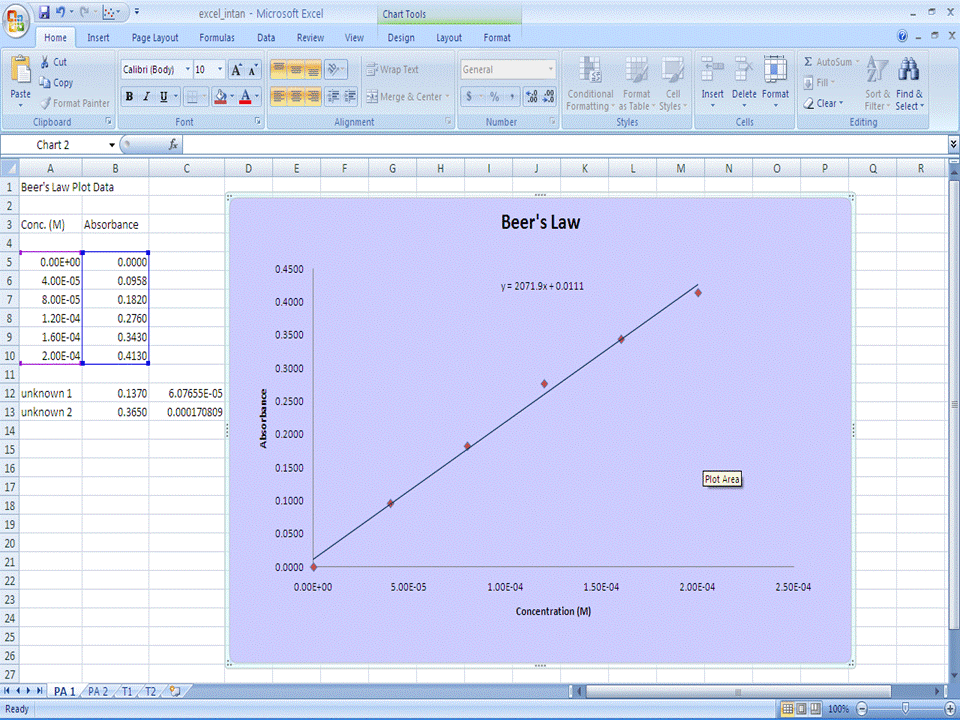
Part 1 - Beer's Law Scatter Plot and Linear Regression
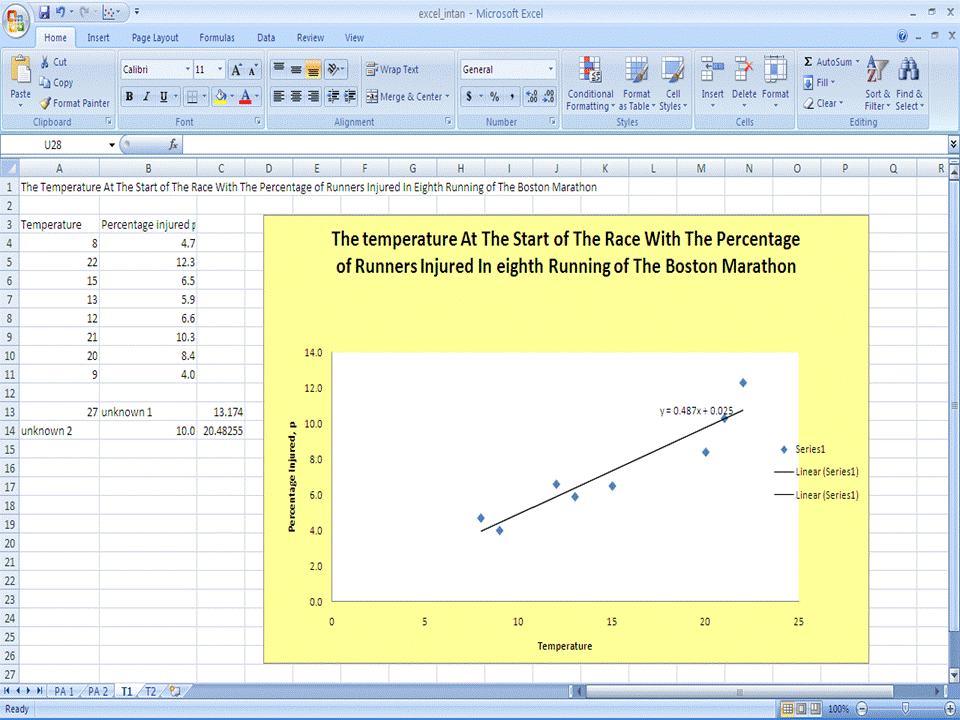
Part 2 - Titration Data Plotting


- Finding the line of best fit
- Quadratic regression

 |
| Energy of Reaction Diagram |
 |
| p-orbital, d-orbital, pi-type orbital |
 |
| Vacuum Distillation Apparatus |
 |
| Two-chain DNA Strand |
 |
| Lipid |
| HTML COLOUR CHART |
| COLOUR | HTML CODE |
| blue | #0000FF |
| Black | #000000 |
| Red | #FF0000 |
| White | #FFFFFF |
| Green | #008000 |
| Purple | #800080 |
| Yellow | #FFFF00 |
| Orange | #FFA500 |
| Violet | #EE82EE |
| Silver | #C0C0C0 |
| Gold | #FFD700 |
| Gray | #808080 |
| Pink | #FFCOCB |
| Fuscia | #FF00FF |
| light blue | #ADD8E6 |
| Sky blue | #87CEEB |
| Aqua | #00FFFF |
| Khaki | #F0E68C |
 | ||
| HtrA |
 |
| LonA |
 |
| ClpP |



